Синдром ангельмана: клинические проявления и подходы к терапии
Содержание:
- Перспективы развития
- Причины
- Что такое Синдром Ретта?
- Причины
- Генетические мутации повышают риск появления заболевания
- Симптомы
- Перспективы развития
- Лечение синдрома Ангельмана
- Симптомы синдрома Ангельмана
- Лечение
- Синдром счастливой куклы
- Причины заболевания
- Съёмочная группа
- Когда мир придуманный желаннее реального
- Схожие с синдромом Аарскога расстройства
- Патогенез
Перспективы развития
Дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарным запасом около 5-10 слов. При этом люди с синдромом Ангельмана любят общаться с другими людьми, играть, как правило, они дружелюбны.
Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам, направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты.
Перспективы развития зависят от степени поражённости хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы).
С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки. Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые — и ночью. Некоторые люди с синдромом Ангельмана способны есть при помощи ножа и вилки, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний». Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом.
Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом.
Причины
Синдром Ангельмана – врожденное заболевание, в основе которого лежит генетический дефект. Патология развивается внутриутробно при делении хромосом. Нарушения в строении 15 хромосомы, переданной от матери, вызывают развитие болезни. Хромосома мутирует в результате делеции — выпадении генов, дупликации — появлении лишних генов, инверсии — обратном расположении генов, инсерции — изменении месторасположения генов, транслокации — присоединении участка одной хромосомы к другой.
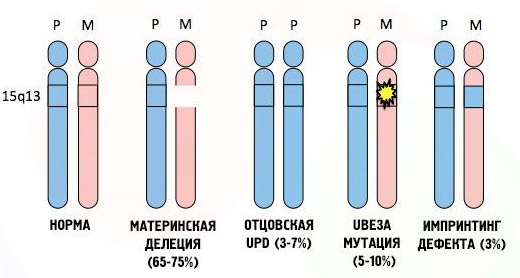
- Дупликация хромосомы является причиной особой формы синдрома, несовместимой с жизнью. Если больные не умирают в младенчестве и достигают половой зрелости, детей они не имеют.
- Делеция хромосомы — причина тяжелой умственной отсталости. Больные дети не могут ходить и говорить, страдают от частых приступов эпилепсии, интенсивность которых имеет крайнюю степень выраженности.
- Мутация одного гена вызывает самую легкую форму синдрома, при которой больные дети могут себя обслуживать и общаться в коллективе. При этом они все равно отстают в развитии от своих сверстников.
Синдром Ангельмана возникает спонтанно. Любой ребенок может появиться на свет с данным недугом. Риск рождения значительно повышается в семьях, где у одного из родителей имеется такой же синдром или уже есть больной ребенок. У абсолютно здоровых родителей могут родиться больные дети. Мутация генов происходит под влиянием факторов, оказывающих тератогенное воздействие на плод:
- Вредные привычки беременной женщины,
- Всплеск эмоций, тяжелые душевные переживания, частые конфликты,
- Воспалительные заболевания органов женской репродуктивной системы,
- Длительный прием психотропных лекарств, наркотических веществ,
- Ионизирующее излучение.
Что такое Синдром Ретта?
Синдром Ретта – является наследственным заболеванием, психоневрологического генеза. Развивается в основном у девочек и приводит к развитию умственной отсталости.
Данный синдром впервые описал невролог из Австрии Андреас Ретт.
Особенности патологии заключаются в том, что ребенок до 18 месяцев развивается хорошо, но затем у ребенка начинают исчезать приобретенные, раннее, навыки: нарушается речь, двигательная активность, предметно-ролевая игра.
Появляются однообразные движения рук, не преследующие какой-либо цели. Нередко ребенок их потирает и заламывает.
Андреасом Реттом были обследованы две девочки, у которых он отметил регрессивный тип психоневрологического расстройства, проявляющийся движениями рук, напоминающих мытье.
Причины
Наши клетки содержат 23 пары хромосом. Каждая пара содержит копии хромосом от отца и от матери. Хромосома 15 содержит ген UBE3A, который отличается тем, что он функционирует в клетках мозга и только на хромосоме материнского происхождения. Копия от отца неактивна.
Синдром Ангельмана обусловлен отсутствием или неисправностью этого гена на материнской хромосоме, что нарушает неврологическое развитие.
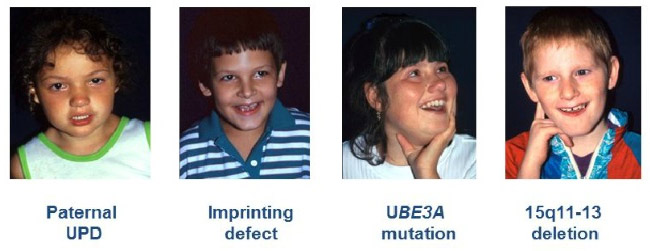
Существует четыре типа генетических аномалий:
- Потеря хромосомы (около 70% случаев): случайная потеря небольшой области «15q 11-12» 15ой хромосомы материнского происхождения, содержащей ген UBE3A.
- Мутация гена UBE3A (около 20% случаев).
- Хромосома 15 отцовского происхождения (от 3 до 5% случаев): обе хромосомы исходят от отца, обе копии гена UBE3A неактивны.
- Изолированная аномалия отпечатка (от 3 до 5% случаев): 15ая хромосома материнского происхождения присутствует, но не имеет отпечатка, который идентифицирует ее как таковую. Поэтому ген UBE3A неактивен.
Также было выдвинуто еще одно негенетическое происхождение патологии. Это характеризуется аномалиями в формировании яйцеклетки, во время размножения или во время эмбрионального развития.
Генетические мутации повышают риск появления заболевания
Синдром Ангельмана встречается нечасто — примерно 1 случай на 10 – 20 тыс. новорожденных малышей. Основной причиной патологии является потеря в 15 хромосоме копий нормальных материнских генов, что происходит по причине нарушения деления данной хромосомы. Кроме того заболевание может возникнуть в связи с мутацией отцовских генов, отцовской дисомией или трисомией.
В норме здоровый человек получает от матери и отца по одной копии 15 хромосомы. Если ребенок получил от кого-то из родителей генетически измененную копию (особенно от матери, так как у нее копии генов более сильные, чем у отца), у ребенка развивается синдром Ангельмана.
Заболевание носит имя Гарри Ангельмана, британского врача-педиатра, который впервые дифференцировал данный синдром в 1965. Тогда он получил название синдрома счастливой марионетки, однако сегодня этот термин не используется, так как это было признано пренебрежительным.
Доктор Ангельман занимался лечением нескольких детей со сходными симптомами и предполагал наличие общего диагноза. Доказать диагноз и получить точные данные на тот момент было не возможно, ввиду отсутствия технологий, которые доступны сегодня. Свои догадки врач отразил в статье под названием «Дети марионетки».
Появление синдрома Ангельмана связано с наличием у родителей будущего ребенка различных хромосомных аномалий. Среди таких отклонений обычно называют:
- трисомию хромосом – присутствие одной или нескольких лишних хромосом в хромосомном наборе;
- инверсию – разворот одного из участков хромосомы на 180 градусов, при этом часть хромосомы пропущена, а гены располагаются в противоположном порядке;
- микроделецию, которая является результатом перестройки Y-хромосомы и обмена участками между хромосомами, наблюдается небольшое количество хромосом, а также может отсутствовать один из генов;
- делецию – нехватку одного из участков хромосомы;
- транслокацию – перенос или присоединение участка одной хромосомы к другой хромосоме;
- дупликацию – копирование части хромосом, результатом чего становится лишний генетический материал;
- кольцевую хромосому – на концах хромосомы отсутствует генетический материал, при этом новообразованные концы соединяются в виде кольца.
Генные мутации, которые могут вызвать развитие синдрома
Рассматриваемая болезнь обязана своим названием врачу-педиатру Гарри Ангельману, который впервые диагностировал отклонение в 1965 году и назвал пациентов кукольными детьми.
Именно картина «Мальчик-марионетка» подарила специалисту уверенность в собственной правоте. Картина отображает смеющегося мальчика, который очень напомнил Гарри его пациентов и он решает написать о них общую статью, которая в дальнейшем получила название «Дети-марионетки».
Признаки синдрома Ангельмана
В 1965 году издается научная работа, интерес о которой был забыт до восьмидесятых, когда упоминания о патологии появились в медицинской среде. Закономерность отсутствия части 15-й хромосомы была установлена только в 1987 году. Поскольку диагноз «синдром счастливой марионетки» унижал и пугал многих родителей, было решено использовать фамилию Ангельмана. Среди современных ученых, изучающих данное явление, стоит выделить М. Б. Миронова и К. Ю. Мухина.
В качестве факторов риска и провоцирующих элементов стоит рассмотреть родительские хромосомные аномалии.
- Кольцевая хромосома – удаление генетического материала с концов хромосомы и соединение новообразованных концов в кольцо.
- Дупликация — частичное повторение хромосом и соответствующее появление лишнего генетического материала.
- Транслокация – присоединение к хромосоме элемента другой хромосомы.
- Делеция предполагает отсутствие одного из секторов хромосомы.
- Микроделеция является следствием перестройки Y-хромосомы и обмена секторов между половыми хромосомами при мейозе. Немного хромосом при этом отсутствует, чаще всего одного из генов тоже нет.
- Инверсия – разворот на 180 градусов одного из участков хромосомы, порядок расположения генов обратный, а часть хромосомы опущена.
- Трисомии хромосом обусловлена наличием одной или больше лишних хромосом в наборе. Причиной такого генетического дефекта является нерасхождение хромосом при делении.
Симптомы
Первые признаки синдрома Ангельмана появляются к концу первого года жизни ребенка. Более явными симптомы становятся по достижению двухлетнего возраста. Синдром характеризуется полиморфностью клинических проявлений. Больных можно узнать сразу. Такие дети имеют характерный внешний вид.
- Особенности внешнего вида больного ребенка: несоответствие маленькой головы и нормального туловища, большой рот, постоянная улыбка, широкие межзубные промежутки, узкие губы, высунутый широкий язык, неправильный прикус, плоский затылок, гладкие ладони, заостренный подбородок, гипопигментированная кожа, стробизм, сколиоз, ходьба на прямых ногах.
- Нарушение психоэмоционального развития проявляется отставанием в умственном развитии, избыточной суетливостью, дружелюбностью, беспричинным смехом, эйфорией.
- Неврологические симптомы: тремор конечностей, атаксия, дискоординация движений, потеря равновесия, гипотония мышц, расстройство сна, истерия, нарушение речи, гиперактивность и гипервозбудимость, сложности с обучением.
- Двигательные расстройства: слабый контроль за движением языка и его беспричинное высовывание, трудности с глотанием и сосанием, поднятые или согнутые во время ходьбы руки, частое слюнотечение, неуемная жажда, излишне активные жевательные движения, нарушение мелкой моторики, эпилепсия.
Дети с синдромом Ангельмана склонны к невербальному общению: воспринимают устную речь, но не могут высказать свои мысли. Они практически не участвуют в разговоре, поскольку их словарный запас состоит из 10-20 слов, необходимых в быту. У больных присутствуют стереотипии – повторяющиеся взмахи руками, кручения кистями и частое хлопанье в ладоши, а также прочие действия, не характерные для здоровых людей. Шаткая походка сопровождается резкими подергиваниями руками, что делает человека похожим на марионетку. Больные чувствуют себя комфортнее в воде и плохо переносят душные помещения и высокую температуру.
С возрастом клиническая картина болезни значительно изменяется. Судороги и эпиприступы возникают все реже или исчезают совсем. Пациенты становятся более спокойными, у них налаживается сон. Мужчины и женщины с данным синдромом имеют моложавую внешность, скрывающую их истинный возраст. Половое созревание у них происходит вовремя, возможно даже рождение детей.
К симптомам патологии у взрослых относятся нарушения мелкой моторики и недержание мочи. Больные не могут справиться с пуговицами и молниями на одежде, но при этом спокойно пользуются вилкой и ложкой. Они дружелюбные, любящие и ласковые. Ожирение – распространенное отклонение среди взрослых больных, особенно женщин. Люди с синдромом Ангельмана склонны к расстройствам вегетативного и соматического характера. Они плохо переносят жаркую погоду, часто страдают от запоров, пищеводного рефлюкса.
Поскольку заболевание является врожденным, все перечисленные аномалии могут проявиться сразу после родов. Современные методы диагностики позволяют выявить синдром внутриутробно и предотвратить рождение больного ребенка.
Перспективы развития
Причиной возникновения заболевания выступает делеция убиквитин-протеин-лигазы E3A (UBE3A), расположенной в 15-й хромосоме. Обычно гены передаются ребенку от родителей в парной комбинации – одна копия по материнской линии, другая – по отцовской.
Клетки организма, как правило, используют информацию из обеих копий, однако в отдельных генах активируется только одна копия.
В случае с синдромом Ангельмана расстройство возникает, когда копия гена UBE3A, передающегося только по материнской линии, отсутствует либо повреждена. Изредка причиной развития синдрома Петрушки выступает наследование двух копий генетической информации только по отцовской линии, а не от обоих родителей.
В соответствии с различными статистическими данными, заболевание поражает 1 ребенка на число новорожденных от 12,000 до 20,000 младенцев. В большинстве случаев синдром Ангельмана у детей возникает на фоне отсутствия этого состояния в семейном анамнезе.
Дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарным запасом около 5-10 слов. При этом дети/люди с синдромом Ангельмана любят общаться с людьми, играть, как правило, они дружелюбны и милы.
Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам , направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты.
Перспективы развития зависят от степени пораженности хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы).
С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки.
Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые — и ночью.
Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом.
Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
На сегодняшний день еще не изобретен чудо-препарат, который бы помог победить это генетическое заболевание. Однако болезнь Ангельмана предполагает симптоматическую терапию, благодаря которой облегчается состояние пациента. При этом прописывается медикаментозное и немедикаментозное лечение. Синдром Петрушки у детей предусматривает такую терапию:
- Назначаются антиконвульсанты. Чаще прописывают Клоназепам, Конвулекс, Ламотриджин. Такие лекарственные средства сводят к минимуму частоту и интенсивность эпиприступов.
- Прописывают витаминотерапию (элементы групп B, C, D и E). Такое лечение укрепляет иммунную систему организма. Однако оно уменьшает эффективность противоэпилептических средств, поэтому все назначения должен делать доктор.
- Прописывается терапия, направленная на устранение проблем с пищеварительным трактом. Такое лечение предусматривает прием слабительных препаратов (Фитолакса или Сенаде) и пробиотиков (Хилак форте, Бифиформа).
- Назначают снотворные. Чаще пациентам прописывают Дифенгидрамин или Мелатонин.
- Показана гормональная терапия. Такое лечение направлено на коррекцию поведения пациентов. Например, гормон секретин улучшает процесс пищеварения, а окситоцин – увеличивают познавательные способности и память.
- В борьбе с проблемами суставов помогут физиопроцедуры. Назначаться могут парафиновые аппликации, массаж, магнитотерапия, электрофорез, аквагимнастика.
При этом заболевании показана поведенческая терапия, занятия с логопедом, психологом и дефектологом. У детей, у которых диагностирован синдром счастливой куклы, отмечаются серьезные нарушения речи. Некоторые из них имеют ограниченный словарный запас, а другим даже сложно воспроизводить отдельные звуки.
Занятия с такими детками должны быть непродолжительными (не более 30 минут), но регулярными: желательно, чтобы они проводились каждый день. По мнению специалистов, начинать можно с рисования пальчиковыми красками. Во время таких занятий ребенок успокаивается, знакомится с цветовой гаммой и развивает мелкую моторику.
Поскольку дети с синдромом Ангельмана лучше воспринимают информацию, когда она визуализирована, желательно при их обучении использовать карточки и другие наглядные пособия. Например, чтобы объяснить такому ребенку понятия «большой»» и «маленький», можно воспользоваться разборной матрешкой. При первом знакомстве малыша с таким наглядным пособием взрослый должен сам открыть игрушку и каждое свое действие прокомментировать. Затем можно попросить ребенка проделать то же самое. Постепенно задача усложняется: например, малыш должен показывать, где большая матрешка.
Симптомы синдрома Ангельмана
Проявления расстройства в психо-эмоциональной сфере:
- задержка развития, в том числе отсутствие лепета и ползания в период от 6 месяцев до 1 года;
- угнетенная обучаемость;
- отсутствие речи либо минимальный навык говорения;
- немотивированный частый смех/улыбка;
- тип личности можно описать как радостный, экзальтированный.
Симптомы расстройства на соматическом уровне:
- приступы эпилепсии, которые обычно возникают в возрасте от 2 до 3 лет;
- затрудненная ходьба/перемещение, скованные или резкие угловатые движения из-за неспособности контролировать произвольные движения (атаксия);
- необычное поведение, такое как внезапные взмахи рук, ходьба с приподнятыми руками и/или на негнущихся ногах;
- небольшой размер головы с уплощением в затылочной зоне (микробрахицефалия);
- волосы, кожа и глаза светлого оттенка (гипопигментация);
- иногда у детей наблюдаются широкий рот, редкие зубы и высунутый язык. В большинстве случаев черты лица пациентов не отличаются чем-то необычным.
Отдельно следует остановиться на особенностях речевого развития и моторики, характерных для людей с синдромом Ангельмана.
У большинства детей с этим расстройством отмечают крайне скудный словарный запас, который может быть ограничен несколькими словами (до одного-двух десятков). Это обусловливает значительные трудности социализации ребенка, у которого наблюдается синдром Петрушки.
При этом дети, имеющие такое расстройство – как правило – понимают не только простые команды, но и способны усваивать информацию в объеме, который значительно превышает их возможности общения и выражения мыслей.
Что касается моторики, то кроме описанных выше проявлений, дети-«Петрушки» могут часто держать руки приподнятыми и согнутыми в локтях и запястьях, и периодически взмахивать руками в моменты возбуждения или во время ходьбы. Именно эта схожесть с движениями марионетки в купе с их угловатостью и скованностью на фоне частой экзальтированности пациентов стала причиной для названия расстройства синдромом счастливой куклы.
У них может наблюдаться как пониженный мышечный тонус туловища (гипотония), так и увеличенный мышечный тонус конечностей (гипертония), а также аномально преувеличенные рефлекторные реакции (гиперрефлексия). У некоторых детей развивается мелкий тремор рук и ног. Эти нарушения могут проявиться в возрасте от 6 месяцев до 1 года.
Освоение некоторых этапных навыков развития моторики – например, ходьбы – происходит с задержкой.
В случаях относительно легкой формы болезни, дети могут начать ходить в 2-3 года. В более тяжелых случаях прямохождение замедлено и происходит рывками – «ригидно», что тоже напоминает движения марионетки. Некоторые дети не ходят до возраста от 5 до 10 лет. Примерно 10% пациентов не способны перемещаться без посторонней помощи.
Лечение
Синдром Ангельмана — неизлечимая генетическая аномалия. В настоящее время ученые-медики активно разрабатывают эффективные терапевтические методики. Если синдром был диагностирован во время внутриутробного развития, специалисты рекомендуют прервать беременность. Новорожденному больному ребенку необходим тщательный уход, забота и высококвалифицированная терапия.
Симптоматическое лечение помогает облегчить состояние больных с синдромом марионетки. Им назначают лекарственные препараты и немедикаментозные процедуры:
Антиконвульсанты снижают частоту и силу эпиприступов. Больным назначают одновременно несколько противосудорожных средств, поскольку приступам характерны несколько видов судорог. К наиболее популярным препаратам относятся: «Вальпроевая кислота», «Конвулекс», «Ламотриджин», «Клоназепам». Эти средства предупреждают судороги, улучшают настроение и психическое состояние больных.
Витаминотерапия — прием витаминов группы В, С, Д и Е. Лечение витаминами следует проводить только по назначению врача, поскольку они снижают эффективность противоэпилептических препаратов.
Снотворные препараты, улучшающие сон у легковозбудимых пациентов – «Мелатонин», «Дифенгидрамин».
При появлении проблем с пищеварением и стулом больным назначают слабительные средства – «Сенаде», «Слабилен», «Фитолакс», пре- и пробиотики – «Хилак форте», «Линекс», «Бифиформ».
Гормональная терапия показана в крайних случаях, когда невозможно другими способами скорректировать поведение больных
Им вводят гормон секретин, нормализующий пищеварение, положительно влияющий на внимание пациентов, а также окситоцин, улучшающий познавательные способности ребенка, память, поведение. Подобная схема лечения гормонами разработана американскими учеными для лечения детей, больных аутизмом.
При синдроме Ангельмана показана поведенческая терапия, работа с психологом, дефектологом и логопедом.
Физиотерапевтические процедуры помогают справиться с гипотонией мышц и проблемами с суставами
Обычно врачи назначают парафиновые аппликации, электрофорез, магнитотерапию. Увереннее стоять на ногах больным помогает ЛФК, профессиональный массаж, аквагимнастика в прохладной воде.
Чтобы избавиться от гиперсаливации, применяют препараты, угнетающие слюнообразование и оперативные вмешательства, направленные на реимплантацию слюнных протоков.
Средства народной медицины, как и гомеопатические препараты, слабоэффективны в лечении синдрома Ангельмана, но абсолютно безопасны. Сбор на основе пиона, солодки и ряски снижает частоту судорожных приступов, отвар лаванды и водный настой пустырника оказывают общее успокаивающее действие.
Синдром счастливой куклы
Коллин Фаррелл с младшим сыном Генри. Фото: todaysparent.com
В 2003 году у Колина Фаррелла родился сын Джеймс. Почти сразу после рождения стало ясно, что он болен. Сначала врачи говорили о ДЦП и аутизме, в конце концов исследования определили — ребенок страдает синдромом Ангельмана. Это означает глубокую умственную отсталость, трудности с речью и движением, судороги и многие другие осложнения здоровья.
При этом дети выглядят счастливыми, часто смеются и улыбаются. Это тоже симптомы болезни, которую называют еще «синдромом счастливой куклы». Сначала Фаррелл решил не раскрывать публично диагноз, но это не давало ему покоя, он чувствовал вину перед ребенком, как будто скрывает что-то постыдное. А это было не так!
В 2007 году Колин Фаррелл приехал в Шанхай, как представитель Паралимпиады, в которой принимают участие взрослые и дети с ограниченными интеллектуальными возможностями. Именно в Шанхае Колин принял решение публично рассказать о сыне, своей радости и гордости за него.
«Борьба за жизнь ребенка с особыми потребностями порой бывает настолько жестокой, что может в клочья порвать ваше сердце, — говорит Фаррелл. — Но любовь — как иголка с ниткой, штопаешь ею раны, и слезы высыхают».
Причины заболевания
Патология развивается из-за возникновения генетической аномалии, при которой из участка седьмой хромосомы выпадает часть генов. Количество исчезнувших генов колеблется от 25 до 29.
Подавляющее большинство случаев заболевания связаны со спонтанной мутацией в половых клетках матери либо отца.
Также возможен аутосомно-доминантный вариант наследования, когда один из родителей ребенка является носителем аномального гена (при этом у него могут отсутствовать какие-либо отклонения в развитии).

К факторам, увеличивающим вероятность возникновения мутации, относятся:
Работа родителей, связанная с профессиональными вредностями, к которым относится взаимодействие с ядовитыми соединениями и радиоактивным излучением.
Плохая экологическая обстановка. Накапливающиеся в организме вредные вещества способны повлиять на возникновение аномалии.
Вредные привычки родителей. И сигареты, и алкоголь, и наркотики неблагоприятно влияют на состояние половых клеток человека, поэтому риск возрастает
Но важно помнить, что дети с аномалиями рождаются не только у людей алкогольной и наркотической зависимостью: гораздо чаще заболевание возникает без каких-либо видимых причин, и ребенок с синдромом Вильямса может родиться у здоровых родителей, не имеющих вредных привычек.
Если в семье есть случаи заболеваемости синдромом Вильямса, это также повышает вероятность возникновения болезни у последующих поколений.
Съёмочная группа
- Автор сценария: Алёна Алова по роману Дины Рубиной «Синдром Петрушки»
- Режиссёр-постановщик: Елена Хазанова
- Оператор-постановщик: Азиз Жамбакиев
- Художник-постановщик: Наталья Навоенко
- Композитор: Николя Рабеюс
- Санкт-Петербургский симфонический оркестр
- Хореограф: Раду Поклитару
- Директор картины: Надежда Попова
- Со-продюсеры: Ундине Филтер, Томас Крал, Пьер-Андре Тьебо совместно с Антонио Эксакустос, Йозеф Райдингер
- Продюсеры: Илья Гаврилов, Александр Новин, Дмитрий Аронин, Ася Темникова, Елена Бренькова, Анна Качко
- Художественный руководитель: Евгений Миронов
Когда мир придуманный желаннее реального
В фильме сказка переплетается с реальностью. И это отражение сложного внутреннего мира главного героя Пети, которого играет Евгений Миронов.
Петя – носитель звуко-зрительного сочетания векторов. Человек со звуковым и зрительным векторами – обладатель мощнейшего абстрактно-образного интеллекта. В искусстве это очень талантливые люди, способные придавать своим творениям необычайную глубину.
Петя – гениальный кукольник. Его куклы неординарны: они почти живые, каждая из них обладает своим характером. Петр живет в придуманном им мире кукол и не желает соприкасаться с миром реальным. Когда речь идет о семейной жизни, он даже говорит своему другу Борису: «Зачем они вообще нужны – эти дети? Это же банальщина. Мне интереснее заводить кукол». За таким восприятием мира стоит череда детских психологических травм.
В детстве Петя видит, как из окна выбрасывается рыжеволосая женщина. Это отпечатывается в детском сознании подсознательным ужасом – зрительные дети очень впечатлительны. Они рождаются со страхом смерти, их надо оберегать от подобных эпизодов. Их огромный эмоциональный потенциал нуждается в развитии, выведении наружу, в любовь и сострадание. У Пети нет таких условий. Он предоставлен сам себе.
Его родителям не до него. Они все время скандалят и кричат. Для ребенка со звуковым вектором, от природы обладающим очень чувствительным слухом, это настоящая травма.
В таких условиях звуковой ребенок все больше и больше отгораживается от мира, погружается внутрь себя, вплоть до аутизма. Однако Петя находит выход в игре в куклы. Зрение помогает звуку выжить в этом мире. Петр оживляет кукол, разговаривает с ними, создает вместе с ними параллельный мир, основанный на его богатой фантазии. Мир, который живет по законам, понятным для ребенка, и защищает его от грубого и несправедливого внешнего воздействия.
Детское спасительное увлечение перерастает в профессию, которая также полностью захватывает все его сознание и время. Однако есть одна ниточка, которая постоянно связывает его с реальностью и не дает полностью уйти в вымышленный мир кукол – это любовь к рыжеволосой Лизе.
Схожие с синдромом Аарскога расстройства
Симптомы следующих расстройств могут быть схожими с таковыми при синдроме Аарскога. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Нунана является относительно распространенным генетическим заболеванием, характеризующимся:
- низким ростом
- дисморфными чертами лица
- врожденным пороком сердца.
Расстройство характеризуется широким спектром симптомов и физических особенностей, которые сильно различаются по степени тяжести. У многих пораженных людей связанные нарушения включают:
- отличительный внешний вид лица;
- широкая или перепончатая шея;
- низкая задняя линия роста волос;
- типичная деформация груди
- низкий рост.
Характерные аномалии головы и лица (черепно-лицевой) области могут включать:
- широко расставленные глаза (глазной гипертелоризм);
- кожные складки, которые могут покрывать внутренние углы глаз (эпикантальные складки);
- опущение верхних век (птоз);
- маленькую челюсть (микрогнатия);
- подавленный носовой корень;
- короткий нос с широким основанием;
- низко посаженные назад повернутые уши.
Также обычно присутствуют отличительные пороки развития скелета, такие как аномалии грудины (грудины), искривление позвоночника (кифоз и/или сколиоз) и вальгусное колено. Многие дети с синдромом Нунана также имеют проблемы с сердцем, такие как обструкция правильного кровотока из нижней правой камеры сердца в легкие (стеноз легочных клапанов).
Дополнительные аномалии могут включать пороки развития определенных кровеносных и лимфатических сосудов, свертываемость крови и дефицит тромбоцитов, трудности в обучении или умеренную интеллектуальную неспособность, неспособность яичек опускаться в мошонку (крипторхизм) к первому году жизни у пораженных мальчиков и/или другие симптомы и признаки.
Синдром Нунана является генетически гетерогенным состоянием, которое может быть вызвано мутациями ряда генов PTPN11, KRAS, SOS1, RAF1, NRAS, RIT1 и SOS2.
Синдром Робинова — это редкое генетическое заболевание, наследуемое как доминантный и рецессивный признак и характеризующееся:
- невысоким ростом из-за задержек роста после рождения;
- отличительными аномалиями головы и лица;
- дополнительные пороки развития скелета;
- генитальные аномалии.
Черты лица у детей с синдромом Робинова напоминают черты лица восьминедельного плода; в медицинской литературе это состояние часто упоминается как «лицо плода».
Характерные черепно-лицевые особенности могут включать:
- аномально большую голову (макроцефалия) с выпуклым лбом;
- широко расставленные глаза;
- маленький вздернутый нос с ноздрями, расклешенными вперёд;
- затонувший (вдавленный) носовой мост.
Пороки развития скелета могут включать необычно короткие кости предплечья (брахимелия предплечья), аномально короткие пальцы рук и ног, постоянную фиксацию пятого пальца в согнутом положении (клинодактилия), необычно маленькие руки с широкими большими пальцами, порок развития ребра, сколиоз и/или недоразвитие одной стороны костей в средней (грудной) части позвоночного столба (гемивертебра).
Генитальные аномалии, связанные с синдромом Робинова, могут включать аномально маленький пенис (микропенис) и неспособность яичек опускаться в мошонку (крипторхизм) у пораженных мужчин и недоразвитие (гипоплазия) клитора и наружных удлиненных складок кожи с обеих сторон вагинального отверстия (половых губ) у пораженных женщин.
Диапазон и серьезность симптомов варьируются от случая к случаю.
Синдром Робинова является генетически гетерогенным состоянием, которое может быть вызвано мутациями в разных генах, таких как WNT5A, ROR2, DVL3 и DVL1.
Патогенез
Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской хромосоме. Этот ген кодирует производство протеина Е6АР, который представляет собой ферментный компонент сложной реакции деградации белков. Е6АР участвует в процессе образования убиквитина – белка системы протеасом, стимулирующего протеолиз дефектных белковых молекул в нейронах головного мозга.
В норме убиквитин маркирует ненужные (неактивные, нефункциональные) белки с целью инициации их уничтожения. Е6АР обеспечивает закрепление убиквитина на поверхности молекулы белка-мишени. Потом протеасомы расщепляют его на пептидные остатки и на аминокислоты. При синдроме Ангельмана убиквитин не закрепляется на дефектных белках, они скапливаются в нервной ткани мозга, нарушается процесс синаптической передачи. Формируются отклонения, задержки в психическом и моторном развитии.