Фенилкетонурия
Содержание:
- Признаки и симптомы
- Лечение фенилкетонурии
- Как проявляется фенилкетонурия
- По данным портала ЗАЧЕСТНЫЙБИЗНЕСФЕДЕРАЛЬНОЕ КАЗЕННОЕ УЧРЕЖДЕНИЕ «ЦЕНТР ПО ОБЕСПЕЧЕНИЮ ДЕЯТЕЛЬНОСТИ КАЗНАЧЕЙСТВА РОССИИ»По данным портала ЗАЧЕСТНЫЙБИЗНЕС7709895509
- Прогноз и профилактика
- Ответы на часто задаваемые вопросы
- Лечение
- Диагностика
- Диагностика
- Диагностика
- Диагностика Фенилкетонурии (ФКУ) у детей:
- Принципы диагностики
- Как развивается фенилкетонурия
Признаки и симптомы
Младенцы с фенилкетонурией обычно кажутся нормальными при рождении. При раннем обследовании и диетическом лечении у пораженных людей симптомы ФКУ могут никогда не проявляться. Однако нелеченные новорожденные с фенилкетонурией, которым не поставлен диагноз в первые дни жизни, могут быть слабыми и плохо питаться. Другие симптомы фенилкетонурии могут включать рвоту, раздражительность и/или красную кожную сыпь с небольшими прыщиками. Задержка развития может быть очевидна в возрасте нескольких месяцев. Средний IQ (коэффициент интеллекта) детей, не получающих лечения, обычно составляет менее 50. Умственная отсталость при ФКУ является прямым результатом повышенного уровня фенилаланина в головном мозге, который вызывает разрушение жирового покрытия (миелина) отдельных нервных волокон. Он также может вызывать депрессию из-за снижения уровня дофамина и серотонина (нейротрансмиттеров) в мозге.
У нелеченных младенцев с фенилкетонурией обычно необычно светлые глаза, кожа и волосы из-за высокого уровня фенилаланина, мешающего выработке меланина, вещества, вызывающего пигментацию. У них также может быть затхлый или «мускусный» запах тела, вызванный фенилуксусной кислотой в моче или потом.
У некоторых нелеченных пациентов с ФКУ присутствуют неврологические симптомы, включая:
- эпилепсию;
- мышечная ригидность;
- гиперрефлексию;
- тремор.
Не леченные женщины с фенилкетонурией, которые забеременели, имеют высокий риск выкидыша или проблем с ростом плода (задержка внутриутробного развития). У детей женщин с нелеченной фенилкетонурией могут быть аномально маленькая голова (микроцефалия), врожденные пороки сердца, аномалии развития или лицевые аномалии. Между тяжестью этих симптомов и высоким уровнем фенилаланина у матери существует тесная взаимосвязь. В результате все женщины с фенилкетонурией, прекратившие лечение, должны возобновить лечение до зачатия и продолжать его на протяжении всей беременности под контролем метаболического генетика и диетолога.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин
Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека. Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются
Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Как проявляется фенилкетонурия
Новорожденные с этим заболеванием не имеют каких-то специфических клинических проявлений, и только в возрасте 2-6 месяцев у ребенка начинается манифестация заболевания. Как только в организм малыша поступает белок молока (грудного, либо заменителей для искусственного вскармливания), появляются первые, неспецифические, признаки фенилкетонурии – вялость, немотивированное беспокойство, гипервозбудимость, постоянные срыгивания, мышечная дистония, судорожный синдром. Одним из самых ранних признаков рассматриваемого заболевания является упорная рвота, но она нередко воспринимается, как клиническое проявление пилоростеноза.
К шестимесячному возрасту у ребенка явно прослеживается отставание в психомоторном развитии – например, малыш становится менее активным, в какой-то период перестает узнавать окружающих его людей (даже самых близких), никаких попыток сесть или встать на ножки не предпринимает. В этот период состав мочи и пота становится аномальным, что и обуславливает характерный для них запах – «мышиный», аромат плесени. Нередко родители отмечают появление на коже грудничка активного шелушения, экземы и дерматита.
Если дети с диагнозом фенилкетонурия не проходят лечение, то у них будет диагностирована микроцефалия, только после полутора лет начнут прорезываться зубы, четко прослеживается задержка речевого развития, а уже к 3, максимум к 4, годам выявляется глубокая умственная отсталость и полное отсутствие речи (исключение составляют только какие-то невнятные звуки).
Дети с фенилкетонурией отличаются диспластическим телосложнением, у них часто встречаются пороки сердца врожденного характера, могут быть вегетативные дисфункции и хронические запоры. Для такого больного ребенка характерными будут:
- тремор верхних конечностей;
- гиперкинезы;
- поза «портного» (нижние и верхние конечности сильно согнуты в суставах);
- семенящая походка.
Все вышеуказанные клинические проявления фенилкетонурии относятся к классической форме заболевания. При атипичных формах рассматриваемой патологии характерными будут повышенная возбудимость, сухожильная гиперрефлексия, тяжелая степень умственной отсталости. Заболевание неизбежно прогрессирует и уже к 2-3 годам ребенок умирает.
По данным портала ЗАЧЕСТНЫЙБИЗНЕСФЕДЕРАЛЬНОЕ КАЗЕННОЕ УЧРЕЖДЕНИЕ «ЦЕНТР ПО ОБЕСПЕЧЕНИЮ ДЕЯТЕЛЬНОСТИ КАЗНАЧЕЙСТВА РОССИИ»По данным портала ЗАЧЕСТНЫЙБИЗНЕС7709895509
О компании:
ФКУ «ЦОКР» ИНН 7709895509, ОГРН 1127746046691 зарегистрировано 30.01.2012 в регионе Москва по адресу: 109240, г Москва, площадь Славянская, 4 СТР.1, 4 ЭТ.;КОМ.2-8;10;12-19;23-28;109-117;119-122;122А. Статус: Действующее. Размер Уставного Капитала — руб.
Руководителем организации является: Директор — Ушаков Андрей Сергеевич, ИНН . У организации 1 Учредитель. Основным направлением деятельности является «управление эксплуатацией нежилого фонда за вознаграждение или на договорной основе».
Рейтинг организации:
Высокий
подробнее
ВНИМАНИЕ: для оценки рисков работы с данной организацией рекомендуем отчет
Должная осмотрительность ?
Статус: ?
Действующее
Дата регистрации: По данным портала ЗАЧЕСТНЫЙБИЗНЕС
?
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
30.01.2012
Размещенные вакансии
|
ОГРН ? |
1127746046691 присвоен: 30.01.2012 |
|
ИНН ? |
7709895509 |
|
КПП ? |
770901001 |
|
ОКПО ? |
38275120 |
|
ОКТМО ? |
45381000000 |
Реквизиты для договора
?
…Скачать
Проверить блокировку cчетов
?
Контактная информация 8-96… Посмотреть
?
Отзывы об организации
?: 0 Написать отзыв
Юридический адрес: ?
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
109240, г Москва, площадь Славянская, 4 СТР.1, 4 ЭТ.;КОМ.2-8;10;12-19;23-28;109-117;119-122;122А
получен 05.06.2013
зарегистрировано по данному адресу:
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
Руководитель Юридического Лица ?По данным портала ЗАЧЕСТНЫЙБИЗНЕС
ДиректорПо данным портала ЗАЧЕСТНЫЙБИЗНЕС
Ушаков Андрей Сергеевич
| ИНН ? |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС |
| действует с | По данным портала ЗАЧЕСТНЫЙБИЗНЕС 17.10.2014 |
Учредители ? ()
|
По данным портала ЗАЧЕСТНЫЙБИЗНЕС По данным портала ЗАЧЕСТНЫЙБИЗНЕС ФЕДЕРАЛЬНОЕ КАЗНАЧЕЙСТВО (КАЗНАЧЕЙСТВО РОССИИ) По данным портала ЗАЧЕСТНЫЙБИЗНЕС 30.01.2012 , ИНН |
Основной вид деятельности: ?По данным портала ЗАЧЕСТНЫЙБИЗНЕС
68.32.2 управление эксплуатацией нежилого фонда за вознаграждение или на договорной основе
Дополнительные виды деятельности:
Единый Реестр Проверок (Ген. Прокуратуры РФ) ?
Реестр недобросовестных поставщиков: ?
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
не числится.
Реестр операторов, осуществляющих обработку персональных данных (Данные РКН) ?
| Регистрационный номер: |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС 77-19-016235 от По данным портала ЗАЧЕСТНЫЙБИЗНЕС 16.12.2019 |
| Дата начала обработки: |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС 27.05.2013 |
Лицензии: ?По данным портала ЗАЧЕСТНЫЙБИЗНЕС
Налоговый орган ?
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
Инспекция Федеральной Налоговой Службы № 9 По Г.москве
Дата постановки на учет: По данным портала ЗАЧЕСТНЫЙБИЗНЕС
30.01.2012
Регистрация во внебюджетных фондах
| Фонд | Рег. номер | Дата регистрации |
|---|---|---|
|
ПФР ? |
087105047126 |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС 08.02.2012 |
|
ФСС ? |
770805598577061 |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС 30.06.2017 |
Коды статистики
|
ОКАТО ? |
45286580000 |
|
ОКОГУ ? |
1327035 |
|
ОКОПФ ? |
75104 |
|
ОКФС ? |
12 |
Финансовая отчетность ФКУ «ЦОКР» ?
|
В качестве Поставщика: , на сумму |
|
В качестве Заказчика: , на сумму |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
Судебные дела ФКУ «ЦОКР» ?
|
найдено по ИНН: По данным портала ЗАЧЕСТНЫЙБИЗНЕС |
|
Ответчик: По данным портала ЗАЧЕСТНЫЙБИЗНЕС , на сумму: 273 437 425,00 руб. |
|
Истец: По данным портала ЗАЧЕСТНЫЙБИЗНЕС , на сумму: 3 439 807 731,00 руб. |
|
найдено по наименованию (возможны совпадения): По данным портала ЗАЧЕСТНЫЙБИЗНЕС |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
Исполнительные производства ФКУ «ЦОКР»
?
|
найдено по наименованию и адресу (возможны совпадения): По данным портала ЗАЧЕСТНЫЙБИЗНЕС |
По данным портала ЗАЧЕСТНЫЙБИЗНЕС
Лента изменений ФКУ «ЦОКР»
?
Не является участником проекта ЗАЧЕСТНЫЙБИЗНЕС ?
Больше информации об организации — в Премиум доступе
Прогноз и профилактика
Предупредить развитие необратимых повреждений органов ЦНС позволяет ранняя диетотерапия. Для того чтобы лечебное питание было назначено вовремя детям практически сразу после рождения проводится массовый скрининг и при необходимости назначается дополнительная диагностика.
Своевременное проведение элиминационной диеты и неукоснительное соблюдение предписаний врача по лечебному питанию позволяет родителям вырастить полностью здорового ребенка, то есть без отклонений в интеллектуальном, физическом и психическом развитии.
Прогноз течения патологии неблагоприятный при поздно начатой диетотерапии. Если в подростковом возрасте диета расширяется неадекватно или ее соблюдение полностью прекращается, то наблюдается снижение способности к обучаемости, нарушение поведенческих норм, расстройства со стороны психики.
Вот почему большинство психотерапевтов рекомендуют придерживаться элиминационной диеты как минимум до 18 лет.
Риск рождения детей, больных фенилкетонурией, оценивается после обследования супружеских пар в медико-генетических центрах. Обязательно такому обследованию должны подвергаться супруги, у которых уже рожден ребенок с таким заболеванием.
Желательно чтобы при планировании зачатия обследование на генетические патологии проходили пары, имеющие близкое кровное родство.
При атипичных формах ФКУ диетотерапия нужных результатов коррекции расщепления фенилаланина не дает. Прогноз течения таких типов патологии неутешительный – малыши либо гибнут в первые годы рождения, либо у них наблюдается глубокая умственная отсталость.
Фенилкетонурия пока единственное наследственное заболевание, раннее лечение которого предупреждает практически на 100 процентов вероятность развития тяжелых осложнений в будущем.
Поэтому родителям нельзя отказываться от неонатального скрининга их малыша в роддоме, а при рождении ребенка дома нужно его обследовать в течение первых трех недель жизни.
https://youtube.com/watch?v=pww9ml5pywE
Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Как выглядят больные фенилкетонурией?
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Какие смеси использовать для ребенка с фенилкетонурией?
витаминымикроэлементыДля детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Какие бывают типы фенилкетонурии?
фенилкетонурии
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
Существует ли профилактика фенилкетонурии?
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Лечение
Единственным методом лечения фенилкетонурии является своевременное налаживание диетического питания, которое необходимо с первых дней. Его суть заключается в ограничении фенилаланина, который содержится в некоторых продуктах питания. Поэтому необходимо строго исключить все белковые продукты. Учитывая биологические процессы в организме, полное исключение фенилаланина из рациона может привести к истощению организма. Поэтому такой больной должен в обязательном порядке получать определенные аминокислотные смеси и белковые гидролизаты и соблюдать диету как минимум 5 лет.
Когда в крови больного снижается концентрация фенилаланина, постепенно в рацион вводят продукты животного происхождения. Также можно добавлять разнообразные фрукты и сезонные овощи, животные и растительные жиры, а также углеводы. При фенилкетонурии лечение предполагает постоянный контроль содержания фенилаланина в крови. Организму малыша необходимо быть снабженным этой аминокислотой в необходимом объеме для его нормального развития и роста.
При соблюдении рекомендаций врачей, как правило, ребенок сможет перейти к обычной системе питания.
Лечение заболевания медицинскими препаратами направлено на устранении симптомов. Например, при фенилкетонурии прописываются лекарства, ориентированные на устранение судорог, а также такие, которые стимулируют интеллектуальную деятельность. Необходимым является назначение курса массажа и лечебной физкультуры. Также могут предлагаться уроки на развитие логики.
Поскольку фенилкетонурия наследуется, то данное заболевание находится в компетенции врача генетика. Поэтому, его диагностирование осуществляется генетиком и педиатром на основании необходимых исследований в комплексе с общей симптоматикой. А в лечении основную роль играет соблюдение правил питания.
Согласно статистике, фенилкетонурией заболевает один ребенок из 10 тысяч новорожденных. С целью профилактики, будущим родителям стоит пройти медико-генетическое консультирование. Как правило, в группе риска пребывают те семьи, у которых были родственники, страдающие от данного заболевания.
Диагностика
Фото: aidsmap.com
Во всех родильных домах проводится массовое тестирование детей на наличие повышенного содержание фенилаланина в крови. Исследование проводится на четвёртый день жизни. Материалом для тестирования служит кровь, взятая из пятки новорождённого. Далее из родильного дома биологический материал доставляют в медико-генетическую консультацию, где проводят полное исследование крови на содержание фенилаланина в крови. Используются различные подходы к выявлению заболевания:
- Тест Гатри – в его основе лежит ингибирование бактерий Bacillus subtilis. Действие ингибитора заканчивается при повышенном содержании фенилаланина в крови. Следовательно колонии бактерий начинают размножаться. Для оценки проведённого теста его сравнивают с определёнными стандартными значениями. Чувствительность теста составляет более 99%. Этот метод применяется во многих странах мира как скрининговый метод выявления фенилкетонурии. В Российской Федерации массово данный тест не применяется.
- Хроматография – метод основывается на движении частиц (молекул белков, аминокислот) в электрическом поле и последующем естественном разделении на однородные фракции. Соответственно образуется фракция фенилаланина в определённом месте в сорбенте. Для оценки теста его сравнивают со стандартными показателями. В качестве скринингово метода не применяется.
- Флюриметрия – определение количества фенилаланина в крови методом хроматографии с помощью автоматических флюориметров. Этот метод во многих странах мира применяют как основной метод автоматизированного скрининга фенилкетонурии.
- Тандемная масс-спектрометрия – метод позволяет установить не только количество фенилаланина в крови, но и соотношение фенилаланин/тирозин.
Далее для уточнения диагноза применяют молекулярно-генетические методы диагностики. Они позволяют установить конкретную поломку гена и ,соответственно, дисфункцию конкретного фермента. Это может оказаться решающим моментом в лечении фенилкетонурии.
Диагностика
Медико-генетическое консультирование семьи
Будущим родителям нужно быть особенно внимательными, если в семьях кого-либо из них имели место случаи рождения больных фенилкетонурией детей. Нельзя исключить возможность появления на свет ребёнка с ФКУ, даже если недуг проявился у дальнего родственника. Коварство заболевания кроется в бессимптомном носительстве повреждённого гена абсолютно здоровыми людьми.
Всем парам, у которых имеются подозрения на наличие наследственных болезней в роду, стоит обратиться к врачу-генетику ещё во время планирования малыша. В медико-генетическом центре (МГЦ) с помощью современных методов диагностики можно выявить носительство мутантного гена и рассчитать риск рождения ребёнка с ФКУ и другими генетическими болезнями.
Инвазивные методы диагностики (хорионбиопсии, амниоцентеза, кордоцентеза) могут помочь определить недуг до рождения крохи. При этом исследуется генетический материал, полученный от плода. Данные методы являются травматичными и могут повлечь за собой спонтанное прерывание беременности. Поэтому применение их оправдано лишь при доказанном носительстве мутантных генов у родителей и высоком риске возникновения ФКУ у малыша.
Неонатальный скрининг
Перед выпиской из родильного дома новорождённых малышей массово обследуют на наследственные заболевания
Для этого важного исследования медработник набирает кровь из пяточки малыша и наносит капельки биологической жидкости на фильтровальную часть тест-бланка. Каждая капля крови предназначена для выявления одного из 5 заболеваний: фенилкетонурии, муковисцидоза, врождённого гипотиреоза, адреногенитального синдрома, галактоземии
Неонатальный скрининг – очень важное и необходимое всем детям без исключения обследование. Он проводится совершенно бесплатно и направлен на сохранение здоровья нации
Отказываясь от манипуляции, родители совершают огромную ошибку, ведь симптомы наследственных недугов не всегда заметны у новорождённого малыша. Яркие клинические проявления нередко возникают, когда помочь ребёнку уже сложно, а изменения в организме крохи приняли необратимый характер.
Этот метод является достоверным в выявлении фенилкетонурии, если малыш получает достаточное количество энтерального питания, грудного молока. Поэтому новорождённым, которые находятся в отделении реанимации, анализ проводят позже. Отличаются и сроки постановки пробы у доношенных и недоношенных детей. Малышам, которые появились в срок, рекомендован набор капиллярной крови на 4-е сутки жизни. Проведение исследование родившимся преждевременно крохам откладывается до 7-х суток.
Анализ следует проводить натощак, что обеспечит более точный результат и повысит диагностическую значимость теста. Бланк с капельками крови малыша и паспортными данными родителей отправляется в лабораторию медико-генетической консультации, где проводится биохимическое исследование.
Родителям нужно обратить внимание на правильность заполнения бланка, корректность своих контактных данных. На проведение исследования уходит в среднем 10 дней, семья к этому времени обычно находится дома
В случае обнаружения положительного результата теста, родителям нужно будет в кратчайшие сроки обратиться в медико-генетический центр, а неправильные контактные данные задержат дальнейшее обследование малыша.
Обследование ребёнка в МГЦ
Чтобы подтвердить заболевание у младенца и установить его причину, малышу придётся пройти многоэтапную процедуру обследования. Повторные биохимические анализы помогут подтвердить повышение уровня аминокислоты в крови и принять решение о необходимости диетотерапии. Проводится исследование уровней фенилаланина и тирозина, активность печёночных ферментов, определение продуктов обмена аминокислот в моче.
Затем осуществляется молекулярно-генетическая диагностика, которая позволяет выявить мутацию в гене РАН, ответственном за развитие фенилкетонурии. С помощью данных методов можно определить и бессимптомное носительство заболевания.
Диагностика
При подозрении на ФКУ проводится целый ряд исследований – клинических и биохимических. Схема первичной диагностики базируется на сборе анамнеза
Во время этого процесса важно установить следующие факты:
- наличие родственных связей между родителями больного ребенка;
- присутствие подобного отклонения у близких родственников;
- проявление тревожных симптомов в виде судорог, нарушения мышечного тонуса, экзематозных поражений кожи, гипопигментации волос, кожных покровов, радужки глаза, специфического затхлого запаха урины.
Проводится ряд лабораторных исследований, помогающих подтвердить предварительный диагноз. Так, при ФКУ наблюдается:
- повышение концентрации фенилалалина в крови до 900 мкмоль/л или больше;
- присутствие фенилпировиноградной, фенилмолочной и фенилуксусной кислот в урине;
- положительная реакция при пробе Феллинга.
Диагностика у новорожденных
ФКУ является весьма опасным заболеванием, сопровождающимся тяжелым течением симптоматики. В связи с этим, также с возможными серьезными последствиями, патология относится к группе болезней, рекомендованных ВОЗ к ранней диагностике у новорожденных.
В России ранняя диагностика фенилкетонурии осуществляется за счет детального обследования новорожденных в родильных домах, и поведении анализа крови с целью определения уровня фенилалалина. Забор биоматериала проводится на 4 – 5 день после рождения малыша. Реже исследуется не кровь, а моча новорожденного.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Принципы диагностики
В настоящий момент на территории Росси массово в роддомах проводится неонатальный скрининг – определение наследственных заболеваний, в том числе и ФКУ, на 3-4 день жизни новорожденного.
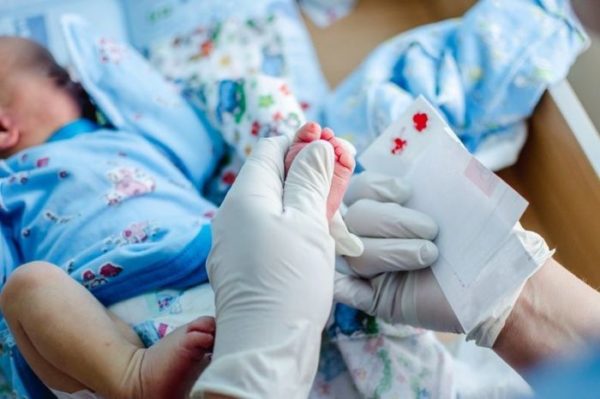
Массовое обследование детей позволяет своевременно выявить фенилкетонурию и назначить соответствующее лечение, что при полном соблюдении терапии исключает дальнейшее прогрессирование патологии.
В случае отсутствия лечения (но не ранее 10 дня жизни новорожденного) в моче выявляются фенилкетоны – продукты расщепления фенилаланина.
Если в крови при проведении неонатального скрининга обнаруживаются данные за развитие патологии, то дальнейшее обследование должно проводиться на базе медико-генетических центров.
Для подтверждения диагноза необходимы следующие исследования:
- Выявление уровня фенилаланина в сыворотке и высохшем пятне крови;
- Определение количества тирозина;
- Копрограмма;
- Потовый тест;
- ДНК-диагностика;
- Хроматография;
- Проба Феллинга – определение в моче фенилпировиноградной кислоты;
- Электроэнцефалография.
В ряде случаев назначается определение показателей фенилаланингидроксилазы в биоптате печени.
Как развивается фенилкетонурия
Врожденное наследственное заболевание, вызываемое нарушением обмена аминокислот, называется фенилкетонурией. Другие названия этой патологии – болезнь Феллинга (по имени впервые описавшего болезнь врача) или фенилпировиноградная олигофрения (основными последствиями прогрессирования заболевания являются тяжелые поражения центральной нервной системы и головного мозга, провоцирующие задержки развития, умственную отсталость).
В России фенилкетонурия регистрируется с частотой 1 случай на каждые 5-10 тыс. новорожденных, в Турции эта пропорция составляет 1:2500, в Японии и некоторых странах Европы – 1:100 000, в странах Африки заболевание практически не встречается. По статистике девочки наследуют патологию в два раза чаще, чем мальчики. В неонатальном периоде клинические проявления болезни отсутствуют, но из-за поступления фенилаланина в организм с пищей манифестация фенилкетонурии происходит в первые полгода жизни.
При болезни Феллинга дефицит печеночного фермента фенилаланин-4-гидроксилазы вызывает нарушения метаболизма фенилаланина, поступающего с белковой пищей. Происходят следующие процессы:
- Запускаются побочные пути окисления фенилаланина, при которых в организме накапливаются его токсичные производные – фенилуксусная, фенилмолочная и фенилпировиноградная кислоты.
- Возникает дефицит тирозина (аминокислота, продукт нормального метаболизма фенилаланина), участвующего в функциях гипофиза, надпочечниках и щитовидной железы, выработке меланина, регулирующего аппетит и процессы жировых отложений.
- Развиваются нарушения обмена липопротеидов, гликопротеидов, белков.
- Происходит расстройство транспорта аминокислот.
- Нарушается обмен катехоламинов и серотонина.
- Образуется избыток фениэтиламина и ортофенилацетата, провоцирующий нарушения липидного метаболизма в головном мозге.
- Разрушение механизма передачи импульсов между клетками нервной системы из-за возникающего дефицита нейромедиаторов.
Причины
Синдром фенилкетонурия у детей в 98% случаев развивается вследствие мутации расположенного на длинном плече 12 хромосомы гена, кодирующего количество фенилаланингидроксилазы (что приводит к недостаточности этого фермента). Генетическая патология носит аутосомно-рецессивный характер наследования, то есть для развития болезни новорожденный должен унаследовать копию дефектного мутировавшего гена и от отца, и от матери, являющихся его гетерозиготным носителем. Этим обусловлена низкая степень распространенности патологии.
Формы и типы
Классическая фенилкетонурия 1 типа, развивающаяся в результате наследования от обоих родителей мутировавшего гена, составляет 98-99% от всех зарегистрированных случаев. Другими, атипичными формами болезни, не поддающимися лечению диетотерапией, но протекающими с аналогичными клиническими проявлениями, являются:
- фенилкетонурия 2 типа, развивающаяся в результате дефицита дегидроптеринредуктазы;
- фенилкетонурия 3 типа, возникающая из-за недостаточности тетрагидробиоптерина.