Иммунодефицит
Содержание:
- Причины и виды первичных иммунодефицитов
- Как заподозрить иммунодефицит?
- Первичные иммунодефициты
- Online-консультации врачей
- Первичные иммунодефициты: причины
- Также в разделе
- Причины иммунодефицита
- Вторичные иммунодецифиты
- Как жить с иммунодефицитом
- Первичные дефициты белков комплемента
- Первичные дефициты клеточного иммунитета
Причины и виды первичных иммунодефицитов
Основным этиологическим факторам этой патологии служит генетический сбой в иммунной системе. В результате чего происходит недостаточное формирование защитных механизмов в организме ребенка. Это приводит к тому, что больной не может противостоять инфекциям, регулярно заболевает различными болезнями. В медицинской практике активно используется классификация первичного иммунодефицита, включающая в себя 5 групп, которые отличаются в зависимости от пораженного звена. Это:
- Гуморальный — связан с нарушением образования антител. Является наиболее распространенным видом. На долю которого приходится порядка 50-60%.
- Клеточный — вызван нарушением дифференцировки и деления лимфоцитов. На его долю приходится не более 10%.
- Комбинированный — гуморальный и клеточный иммунодефицит. Занимает второе место по распространенности, охватывая около 20% больных.
- Иммунодефицит, вызванный недостаточностью фагоцитов. Составляет от 10 до 15% случаев. Нарушает способность фагоцитарный клеток уничтожать патогенные микроорганизмы. Для этой разновидности больше всего характерно появление кожных инфекций, вызванных стафилококками и грамотрицательной микрофлорой.
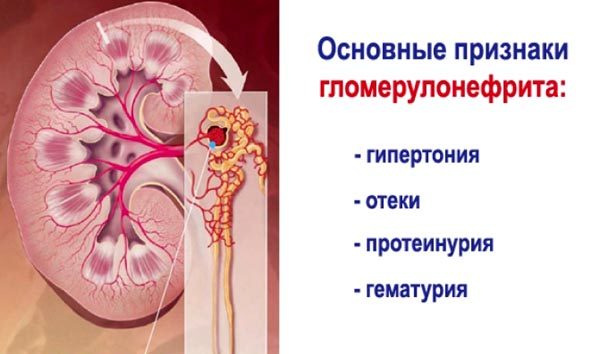
- Комплементарный. Встречается достаточно редко (составляет менее 2%). Включает в себя отдельные расстройства компонентов комплемента. Может иметь как наследственный (бывают аутосомными и рецессивными), так и приобретенный характер. Такие состояния приводят к дефективным опсонизациям, фагоцитозу и лизису патогенетических объектов, равно как и к недостаточной чистке комплексов АГ-АТ. Наиболее серьезными и жизнеугрожающими представителями данной категории служит СКВ и аутоиммунный гломерулонефрит.
Как заподозрить иммунодефицит?
Нередко, особенно у родителей, возникает вопрос: как понять – частые заболевания являются результатом ослабленного иммунитета или же речь идет об иммунодефиците? На что следует обратить внимание? Существует несколько тревожных признаков, при наличии которых лучше посетить иммунолога
- Частые повторения одного и того же заболевания бактериальной природы, например, гнойные отиты, бесконечные диареи, кожные инфекции;
- Инфекционное заболевание протекает в тяжелой форме, несмотря на проводимое лечение, улучшение не наступает в течение длительного времени;
- В ходе обследования при инфекционном заболевании были обнаружены редкие для данной патологии возбудители болезни;
- Инфекции имеют наследственный характер, например, родители так же часто страдали от такого же заболевания;
Для иммунодефицитов характерны тяжелые инфекции с постоянными обострениями, бронхиты, пневмонии, отиты, синуситы, лимфадениты – частые спутники человека с нарушениями иммунитета. Нередко человек страдает кожными заболеваниями: пиодермией, фурункулезом, флегмоной, возможны грибковые поражения, появление герпеса различной локализации. Простудные заболевания нередко сопровождаются стоматитом.
Помимо клинических проявлений подтвердить диагноз можно, пройдя иммунологическое обследование. Скрининговые тесты первого уровня проводятся во многих клиниках, углубленное иммунологическое обследование можно пройти только в учреждении, где есть лаборатория клинической иммунологии. При подозрении на первичный иммунодефицит с помощью анализов можно определить тип мутации, вызвавшей болезнь, и неблагополучное звено в иммунной системе.
Первичные иммунодефициты
Определение и классификация
Первичные иммунодефициты — это врожденные (генетические или эмбриопатии) дефекты иммунной системы. В зависимости от уровня нарушений и локализации дефекта они бывают:
- гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов
- Х-сцепленная агаммаглобулинемия (болезнь Брутона)
- Гипер-IgM синдром
- Х-сцепленная
- аутосомно-рецессивная
- делеция генов тяжелых цепей иммуноглобулинов
- дефицит k-цепей
- селективный дефицит субклассов IgG с или без дефицита IgA
- дефицит антител с нормальным уровнем иммуноглобулинов
- общая вариабельная иммунная недостаточность
- дефицит IgA
- клеточные
- синдром Ди Джоржи
- первичный дефицит CD4 клеток
- дефицит CD7 Т-клеток
- дефицит ИЛ-2
- множественная недостаточность цитокинов
- дефект передачи сигнала
- комбинированные:
- синдром Вискотта-Олдрича
- атаксия-телеангиоэктазия (синдром Луи-Бар)
- тяжелая комбинированная иммунная недостаточность
- Х-сцепленная с полом
- аутосомно-рециссивная
- дефицит аденозиндезаминазы
- дефицит пуриннуклеозидфосфорилазы
- дефицит молекул II класса МНС (синдром лысых лимфоцитов)
- ретикулярная дизгенезия
- дефицит CD3γ или CD3ε
- дефицит СD8 лимфоцитов
- недостаточность системы комплемента
- дефекты фагоцитоза
- наследственные нейтропении
- инфантильный летальный агранулоцитоз (болезнь Костмана)
- циклическая нейтропения
- семейная доброкачественная нейтропения
- дефекты фагоцитарной функции
- хроническая гранулематозная болезнь
- Х-сцепленная
- аутосомно-рециссивная
- дефицит адгезии лимфоцитов I типа
- дефицит адгезии лейкоцитов 2 типа
- дефицит глюкозо-6-дегидроегназы нейтрофилов
- дефицит миелопероксидазы
- дефицит вторичных гранул
- синдром Швахмана
- наследственные нейтропении
Клиническая картина ИДС
Клиника имеет ряд общих черт:
- 1. Рецидивирующие и хронические инфекции верхних дыхательных путей, придаточных пазух, кожи, слизистых оболочек, желудочно-кишечного тракта, часто вызываемые оппортунистическими бактериями, простейшими, грибами, имеющие тенденцию к генерализации, септицемии и торпидные к обычной терапии.
- 2. Гематологические дефициты: лейкоцитопении, тромбоцитопении, анемии (гемолитические и мегалобластические).
- 3. Аутоиммунные расстройства: СКВ-подобный синдром, артриты, системная склеродермия, хронический активный гепатит, тиреоидит.
- 4. Нередко ИДС сочетается с аллергическими реакциями 1 типа в виде экземы, отека Квинке, аллергическими реакциями на введение лекарственных препаратов, иммуноглобулина, крови.
- 5.Опухоли и лимфопролиферативные заболевания при ИДС встречаются в 1000 раз чаще, чем без ИДС.
- 6. У больных с ИДС часто отмечаются расстройства пищеварения, диарейный синдром и синдром мальабсорбции.
- 7. Больные с ИДС отличаются необычными реакциями на вакцинацию, а применение у них живых вакцин опасно развитием сепсиса.
- 8. Первичные ИДС часто сочетаются с пороками развития, прежде всего, с гипоплазией клеточных элементов хряща и волос. Кардиоваскулярные пороки описаны, главным образом, при синдроме Ди-Джоржи.
Лечение первичных ИДС
Этиотропная терапия заключается в коррекции генетического дефекта методами генной инженерии. Но такой подход является экспериментальным.
Основные усилия при установленном первичном ИДС направлены на:
- профилактику инфекций
- заместительную коррекцию дефектного звена иммунной системы в виде трансплантации костного мозга, замещения иммуноглобулинов, переливания нейтрофилов.
- заместительную терапию ферментами
- терапию цитокинами
- витаминотерапию
- лечение сопутствующих инфекций
- генная терапия
- иммуномодулирующя терапия
В 2018 году российский препарат на основе высокоочищенных прошел . В ходе испытаний была подтверждена безопасность применения лекарственного средства. Планировалось, что после регистрации и завершения дополнительных исследований, препарат возможно будет применять в качестве заместительной и иммуномодулирующей терапии у пациентов со сниженным или отсутствующим уровнем синтеза антител. Средство направлено на обеспечение нормализации уровня иммуноглоублина до оптимальных значений и повышение сопротивляемости организма к патогенам.
Online-консультации врачей
| Консультация эндоскописта |
| Консультация дерматолога |
| Консультация детского невролога |
| Консультация онколога-маммолога |
| Консультация общих вопросов |
| Консультация специалиста по лечению за рубежом |
| Консультация гастроэнтеролога детского |
| Консультация генетика |
| Консультация трихолога (лечение волос и кожи головы) |
| Консультация пластического хирурга |
| Консультация уролога |
| Консультация стоматолога |
| Консультация косметолога |
| Консультация массажиста |
| Консультация сосудистого хирурга |
Новости медицины
Футбольные фанаты находятся в смертельной опасности,
31.01.2020
«Умная перчатка» возвращает силу хвата жертвам травм и инсультов,
28.01.2020
Назван легкий способ укрепить здоровье,
20.01.2020
Топ-5 салонов массажа в Киеве по версии Покупон,
15.01.2020
Новости здравоохранения
Глава ВОЗ объявил пандемию COVID-19,
12.03.2020
Коронавирус атаковал уже более 100 стран, заразились почти 120 000 человек,
11.03.2020
Коронавирус атаковал 79 стран, число жертв приближается к 3200 человек,
04.03.2020
Новый коронавирус атаковал 48 стран мира, число жертв растет,
27.02.2020
Первичные иммунодефициты: причины
Первичные иммунодефициты относят к редким заболеваниям, в которых до сих пор отсутствуют точные данные о распространенности более чем 170 различных молекулярно-генетических дефектов.
Чаще всего иммунодефицит связан с недостатком иммуноглобулина А (IgA), белка-индикатора, который отвечает за защиту организма от инфекций – 30%.
Затем следует дефицит подкласса IgG – всего 20 % иммуноглобулина, который является доминирующим среди всех сывороточных иммуноглобулинов.
Затем следует гипогаммаглобулинемия (23%) – заболевание, возникающее в результате дефицита β-клеток (лимфоцитов) в сочетании с уменьшением размера антител-иммуноглобулинов.
Другие недостатки встречаются реже, например сочетание нарушения дефицита β и Т-лимфоцитов (11%), дефицит фагоцитов (8%) и фактора комплемента (3%).
Общая переменная иммунодефицита является наиболее распространенным симптоматическим иммунодефицитом в организме человека.
Заболевание характеризуется низкой концентрацией иммуноглобулина в сыворотке, дефектной продукцией специфических антител и повышенной подверженностью бактериальной инфекции.
Люди чаще обычного болеют респираторными инфекциями дыхательных путей, сопровождающихся аутоиммунной ацитопенией – пониженным содержанием форменных элементов в крови, вызванных аутоиммунной реакцией.
А также лимфопролиферативным синдромом (группа заболеваний, морфологическим субстратом которых являются клетки лимфоидной природы), и гранулемой – поражением тканей организма.
Селективный иммуноглобулин А характеризуется низким 0,05 г/дл или не детектируемым уровнем сывороточного IgA у детей до 4 лет с нормальным уровнем IgG и интактным (неповрежденным, не вовлеченным в какой-либо процесс) иммунным ответом.
Дефицит подкласса IgG может быть изолирован или связан с другими четко определенными первичными иммунодефицитами, которые характеризуются нормальными уровнями общего IgG в сыворотке с пониженными уровнями одного или нескольких подклассов IgG.
Диагностика первичного иммунодефицита и нарушений, связанных с дефицитом антител
Распространенность хронического риносинусита (воспаление слизистой носа и околоносовых пазух) составляет 36-78%.
Примерно в 41% случаев диагностируют острый рецидивирующий или хронический риносинусит.
Лабораторный параметр, указывающий на дефицит иммуноглобулина, представляет гуморальный иммунный ответ на пневмококковую вакцину. Люди со сниженным титром антител к пневмококковой вакцине страдают от риносинусита в 77% случаев.
Около 20% педиатрической популяции имеют дефицит подкласса IgG без каких-либо клинически явных симптомов. С другой стороны, около 90% детей с дефицитом IgA клинически симптоматичны.
Предполагаемый диагноз первичных иммунодефицитов при рецидивирующих инфекциях должен быть подтвержден титрами патологических антител.
В литературе описана задержка диагностики, в которой между первым проявлением и окончательным диагнозом может пройти период 4,7-15 лет.
Рентгенологическое исследование с КТ может помочь в постановке диагноза. От 53% до 90% взрослых и детей имеют все признаки хронического синусита при компьютерной томографии.
Люди с хроническим риносинуситом хорошо реагируют на медикаментозную терапию с внутривенным введением иммуноглобулина. Продолжительность терапии -12 месяцев.
В процессе лечения постепенно снижается употребление наркотических препаратов, сокращаются эпизоды риносинусита с 9 до 4 в год. КТ показала улучшение местных результатов.
Дети с хроническим риносинуситом и отсутствием реакции на медикаментозную терапию требуют дополнительного исследования на предмет возможного синдрома дефицита антител.
И все же, у 54-63% пациентов с сердечнососудистыми заболеваниями даже после внутривенной терапии был хронический риносинусит. Исследования мазка показали положительную бактериальную или вирусную культуру, несмотря на внутривенное введение иммуноглобулина G.
Терапевтический эффект при хроническом риносинусите не доказан, а хирургическое вмешательство в терапии первичных иммунодефицитов до настоящего времени не изучалось.
С этой статьей читают:
Синдром Марфана: симптомы
Также в разделе
| Дискусійні питання малюкової кольки (у світлі Римських критеріїв ІV) В.Є. Хоменко , лікар педіатр вищої категорії МЦ Eurolab , к.мед.н., доцент, кафедра педіатрії №2 Національного медичного університету імені О.О. Богомольця, м…. | |
| Лямблиоз. Клинические проявления лямблиоза у детей. Диагностика. Лечение лямблиоза. Лямблиоз — широко распространенное заболевание человека, которое вызывается микроорганизмами из семейства простейших – Protozoe Giardia Lamblia (жиардии лямблии)…. | |
| Хронический гастрит у детей. Симптомы. Диагностика. Лечение. Хронический гастрит — длительно существующее воспаление слизистой оболочки желудка диффузного или очагового характера с постепенным развитием её атрофии и… | |
| Пролапс митрального клапана у детей Пролапс митрального клапана — прогибание одной или обеих створок митрального клапана в полость левого предсердия во время систолы левого желудочка. Это одна из… | |
| Лимфатико-гипопластический диатез. Причины возникновения. Симптомы. Диагностика. Лимфатико-гипопластический диатез — аномалия конституции, сопровождающаяся диффузной гиперплазией лимфоидной ткани (генерализованное увеличение… | |
|
Рахитоподобные заболевания Рахитоподобные заболевания отличаются от рахита этиологией, но имеют фенотипическое сходство с ним (деформации скелета), в основе которого лежит… |
|
| Смешанное вскармливание. Питание детей старше года. Режим питания детей старше года. При недостаточности молока у матери вводят докорм теми же молочными смесями, что и при искусственном вскармливании. Сначала ребёнку дают грудь и только после… | |
| Часто болеющие дети Часто болеющие дети – это не нозологическая форма и не диагноз, а только группа диспансерного наблюдения, включающая детей с частыми респираторными… | |
|
Острый облитерирующий бронхиолит. Причины возникновения. Симптомы. Диагностика. Лечение. Острый облитерирующий бронхиолит — распространённое поражение эпителия бронхиол с последующей организацией экссудата и гранулематозной реакцией, а… |
|
| Рахит. Причины возникновения. Симптомы. Диагностика. Лечение. Профилактика. Рахит — полиэтиологичное обменное заболевание, обусловленное несоответствием между высокой потребностью растущего организма в солях фосфора и кальция и… |
Причины иммунодефицита
По этиологии различают первичные и вторичные иммунодефициты.
Первичные иммунодефициты развиваются на фоне генетических нарушений. При этом происходит нарушение отдельных составляющих иммунитета:
Гуморального ответа:
Болезнь Брутона;
- Общий вариабельный иммунодефицит;
- Селективный дефицит иммуноглобулинов;
- Транзиторная гипогаммаглобулинемия у детей.
Клеточного звена:
- Хронический слизисто-кожный кандидоз;
- Синдром Ди Джорджи.
Системы фагоцитов:
- Синдром Чедиака – Стейнбринка – Хигаси;
- Хронический гранулематоз;
- Синдром Джоба;
- Дефицит экспрессии молекул адгезии.
Комплимента: врожденный ангионевротический отек.
Выделяют также комбинированные иммунодефициты:
- Тяжелый комбинированный иммунодефицит;
- Синдром Луи-Бар;
- Комбинированный иммунодефицит с повышенным иммуноглобулином М;
- Иммунодефицит с карликовостью;
- Синдром Вискотта-Олдрича.
Первичный иммунодефицит сопровождает человека в течение всей жизни. Умирают такие пациенты от инфекционных осложнений.
Вторичный иммунодефицит развивается по причине воздействия на организм различных инфекций и неблагоприятных факторов внешней среды. Вторичные иммунодефициты (кроме вируса иммунодефицита человека) хорошо поддаются лечению и являются обратимыми.
В качестве основных причин вторичных иммунодефицитов выступают:
- Хронические вирусные и бактериальные инфекции, паразитарные инвазии (вирус иммунодефицита человека, стафилококкоз, туберкулез, пневмококкоз, токсоплазмоз, аскаридоз, малярия, лейшманиоз и другие). При инфекционных хронических заболеваниях иммунная система сильно изменяется, происходит интоксикация организма, угнетается функция кроветворения. При наличии вируса иммунодефицита человека иммунодефицит опосредуется поражением клеток иммунной системы;
- Нарушение питания и истощение организма. Иммунитет человека особенно чувствителен к недостатку минералов, витаминов, питательных веществ. Поэтому снижение защитных сил организма чаще наблюдается в период сезонной витаминной недостаточности;
- Диарейный синдром;
- Гельминтозы;
- Тяжелые травмы и операции. Любое серьёзное заболевание или оперативное вмешательство ведет к вторичному иммунодефициту. Это связано с интоксикацией организма, нарушением метаболизма, а также производством организмом большого количества гормонов надпочечников, угнетающих функцию иммунной системы;
- Стресс-синдром;
- Большие потери крови, ожоги, заболевания почек;
- Эндокринопатии (гипертиреоз, гипотиреоз, сахарный диабет);
- Острые и хронические отравления токсическими веществами, наркотическими средствами, лекарственными препаратами. Сильно снижают иммунитет цитостатики, антибиотики, глюкокортикоиды, антиметаболиты;
- Низкая масса тела при рождении;
- Злокачественные новообразования;
- Аутоиммунные заболевания;
- Старческий и детский возраст, беременность. Снижение иммунитета в этих случаях обусловленофизиологическими особенностями организма в эти периоды жизни человека.
Вторичные иммунодецифиты
Это нарушения системы иммунитета, которые развиваются как у детей, так и у взрослых. Считается, что данный вид иммунодефицита у детей не зависит от генов родителей.
Три формы вторичных иммунодефицитов
- приобретенная
- спонтанная
- индуцированная
Среди приобретенных форм известен СПИД. Индуцированный иммунодефицит спровоцирован некой причиной, к примеру, действием цитостатиков, кортикостероидов, рентгеновским облучением, хирургическими операциями и травмами. Индуцированные формы вторичного иммунодефицита у детей считаются проходящими – то есть устранение причины приводит к ликвидации состояния иммунодефицита.
Спонтанная форма не имеет видимой причины. У ребенка проявляются повторяющиеся инфекционно-воспалительные процессы бронхолегочного аппарата и придаточных пазух носа, а также желудочно-кишечного тракта, урогенитальной системы, кожи и мягких тканей, глаз. Спонтанная форма иммунодефицита более частая, чем приобретенная или индуцированная.
При таких процессах оценить состояние иммунитета часто трудно. Поскольку причину и следствие разграничить тяжело. Нарушение системы иммунитета может привести к заболеванию или заболевание может привести к снижению показателей иммунитета. Вторая причина – тяжело получить необходимый для исследования материал, и методические возможности на сегодня очень ограничены.
Выделяют три группы больных детей с вторичным иммунодефицитом:
1. больные с типичными клиническими признаками нарушения системы иммунитета в сочетании с изменениями лабораторных показателей
2. больные с симптомами нарушения системы иммунитета, не подтвержденные лабораторными данными
3. больные с отклонениями в лабораторных данных, но без внешних проявлений иммунодефицита
Чтобы назначить специальные иммуностимулирующие препараты, необходимы уточнения. Иногда у детей падает иммунитет временно, и это не повод назначать терапию. К примеру, на ребенка повлияли стрессовые ситуации или факторы биологической природы. В некоторых случаях требуется постоянное слежение за состоянием иммунного статуса ребенка.
Критерием назначения иммуномодуляторов ребенку является клиническая картинка (внешние проявления). Иммунодефицит проявляется, прежде всего, повышением инфекционной заболеваемости. В таких случаях иммуномодуляторы помогают организму противостоять инфекциям.
Микроорганизмы условно делят на 2 группы: внутриклеточные и внеклеточные. Внеклеточные возбудители подавляются нейтрофилами, которые оказывают бактерицидную и поглотительную функции. Формируют устойчивость к внутриклеточным возбудителям такие клетки как NK-клетки, макрофаги, Т-лимфоциты.
Возбудители заболеваний, попадая в организм ребенка, встречаются, прежде всего, с макрофагами. Ранний ответ организма зависит именно от них. Макрофаги запускают развитие гуморального и клеточного ответа. Потому при иммунодефицитах применяют иммуномодуляторы, действующие в основном на клетки моноцитарно-макрофагальной системы, например, полиоксидоний.
Применяют иммуномодуляторы, которые воздействуют в основном на Т-лимфоциты, которые синтезируют цитокины. Активируется микробицидная и поглотительная функции нейтрофилов и клеток ММС. На Т-систему иммунитета действуют препараты, полученные из вилочковой железы крупного рогатого скота, к примеру, имунофан и Т-активин.
Когда хронические инфекционно-воспалительные процессы обостряются, врачи прописывают детям антибиотики. Часто параллельно принимают также иммуномодуляторы. Антибиотики подавляют действие возбудителя болезни или убивают его, а иммуномодуляторы повышает функциональную активность фагоцитов, усиливая их бактерицидное действие. Эффективны такие иммуномодуляторы как ликопид, полиоксидоний, миелопид, имунофан, Т-активин и проч.
Редко иммуномодуляторы детям назначают без приема других препаратов, как монотерапию. Например, это актуально для случаев существенного угнетения одного из составляющих иммунной системы с клинической манифестацией оппортунистической инфекции.
Когда детям с вторичным иммунодефицитом назначают противогрибковые, антибактериальные или противовирусные препараты, одновременно приписывают иммуномодуляторы с преимущественным способом действия на клетки ММС, к примеру, полиоксидоний.
Как жить с иммунодефицитом
Независимо от формы иммунодефицита, все без исключения пациенты должны избегать контакта с инфекцией: любая для них может оказаться смертельной. Помните: не заразиться невозможно. Конечно, для многих лечение будет пожизненным, скорее всего – дорогостоящим. К тому же семью ожидают постоянные госпитализации, антибиотики, оформление больничных листов – взрослым пациентам или родителям больных детей.
И главное: продолжительность жизни пациентов с врожденной формой зависит от своевременного и регулярного приема медикаментов! Для пациентов с приобретенными формами важно также регулярно проходить обследования для контроля и профилактики резкого прогрессирования. И хотя существует более 250 разновидностей нарушений, приводящих к иммунодефициту, есть люди, для которых сбой в работе иммунной системы и СПИД означают одно и то же
А ведь первичный иммунодефицит не имеет ничего общего со СПИДом, им нельзя заразиться. Но, к сожалению, больным нередко приходится сталкиваться с непониманием
И хотя существует более 250 разновидностей нарушений, приводящих к иммунодефициту, есть люди, для которых сбой в работе иммунной системы и СПИД означают одно и то же. А ведь первичный иммунодефицит не имеет ничего общего со СПИДом, им нельзя заразиться. Но, к сожалению, больным нередко приходится сталкиваться с непониманием.
Кстати, в России для детей, страдающих опасными нарушениями иммунитета, создан благотворительный фонд «Подсолнух». Также существует организация «Общество пациентов с первичным иммунодефицитом» объединяющая больных и членов их семей. Целью организации является защита и поддержка больных, в том числе юридическая, информационная и психологическая.
А знаете, что 90% пациентов с иммунодефицитом в нашей стране погибают, так и не получив помощь? Поздняя диагностика, а то и ее отсутствие, неправильное лечение, дефицит лекарств – наша действительность. Некоторым приходится регулярно проходить курс терапии и соблюдать многочисленные ограничения. А ведь современная медицина может обеспечить многим больным достаточно долгую и полноценную жизнь. Но для этого надо, в первую очередь, не отмахиваться даже от, казалось бы, пустяковых жалоб, и при любых нарушениях обратиться к врачу. Ведь чтобы выявить причину, которая не позволяет иммунной системе нормально функционировать, достаточно обычного клинического обследования.
Оксана Матиаш, врач общей практики
Иллюстрации: Юлия Прососова
Первичные дефициты белков комплемента
Недостаточность белков комплемента проявляется по-разному в зависимости от того, какой (или какие) белки отсутствуют.
Выделяют три группы заболеваний, связанных с первичным дефицитом комплемента:
- Комплемент-зависимые иммунодефицитные синдромы
- Комплемент-ассоциированные аутоиммунные болезни
- Наследственный ангионевротический отёк Квинке—Ослера.
Комплемент-зависимые иммунодефицитные синдромы
Комплемент-зависимые иммунодефицитные синдромы — заболевания, сопровождающиеся недостаточностью антибактериальной защиты организма. Они проявляются частыми инфекционными процессами в различных органах и тканях. Поскольку белки комплемента при активации играют роль хемоаттрактантов и опсонинов, обеспечивая эффективную функцию фагоцитирующих клеток, то при дефиците компонентов комплемента формируется вторичная недостаточность функции макрофагов и нейтрофильных гранулоцитов. Особенно часто инфекционные процессы при этом вызваны стрептококками, в частности пневмококками, и Haemophilus influenzae. В эту группу включают недостаточность С3b-инактиватора, белков С3, С6 и С8.
Недостаточность С3b-инактиватора. С3b-инактиватор играет роль ингибитора альтернативного пути активации комплемента. При его отсутствии происходит быстрое потребление С3-компонента (вторичный дефицит С3), который в нормальных условиях принимает активное участие в антибактериальной защите организма. Белка С3 у больных в плазме примерно 20 % от нормы. Однако на 75 % он представлен С3b-фрагментом. Уровень нативного С3 составляет всего 5 % от нормы. Скорость расщепления С3 у больных повышена почти в 5 раз. Показано, что через 2 часа после инъекции нативного С3 расщеплению подвергается 40 % введённых молекул. Помимо вторичного дефицита С3 формируется вторичная недостаточность белка С5, однако она менее выражена (примерно 40 % от нормального уровня). Заметно снижена концентрация фактора В — 5 % от нормы (расщепление фактора В происходит под влиянием фактора D). Уровень пропердина снижен незначительно. Больные при этом заболевании страдают различными бактериальными инфекциями.
Недостаточность С3. Недостаточность С3-компонента комплемента также проявляется различными бактериозами. В основе заболевания, в отличие от недостаточности С3b-инактиватора, лежит первичный дефицит С3-белка.
Комплемент-ассоциированные аутоиммунные болезни
Недостаточность белков комплемента провоцирует возникновение аутоиммунных заболеваний, прежде всего (1) красной волчанки, (2) так называемого волчаночно-подобного синдрома и (3) ревматоидного артрита. Часто поражаются почки по типу гломерулонефрита. У больных также описаны пурпура Шёнлейна—Геноха и полимиозит. К этим заболеваниям относятся недостаточность белков С1, С2, С4 и С5. Гены этих белков сцеплены с генами иммунного ответа (генами МНС), поэтому дефекты их, как правило, обоюдны.
Недостаточность С2. Недостаточность С2 является самым частым вариантом первичного дефицита белков комплемента. С2 синтезируют фиксированные и блуждающие макрофаги, фагоцитарная функция которых при этом не нарушена.
Наследственный ангионевротический отёк Квинке—Ослера
К третьей группе состояний, связанных с первичной недостаточностью комплемента, относится наследственный ангионевротический отёк Кви́нке—О́слера, в основе которого лежит недостаточность С1-ингибитора. У отдельных больных при этом возникают аутоиммунные процессы, прежде всего красная волчанка.
Первичные дефициты клеточного иммунитета
К первичным дефицитам клеточного иммунитета относятся следующие заболевания:
- Синдром Ди Джорджи
- Синдром Дункана
- Недостаточность пуриннуклеозидфосфорилазы
- Оротацидурия
- Биотин-зависимые ферментопатии.
Синдром Ди Джорджи
В основе синдрома Ди Джо́рджи (Di George) лежит гипоплазия тимуса. Синдром описан в г. Считается, что это заболевание не является наследственным, оно возникает в результате приобретённого нарушения органогенеза в области III—V жаберных дуг (глоточных карманов) на 6—8 неделе беременности. Поэтому, кроме порока тимуса, отмечаются дефекты околощитовидных желёз, сердца и крупных сосудов, а также орофациальные пороки (микростомия, микрогнатия, гипертелоризм, низкое расположение ушных раковин).
Результатом гипоплазии паращитовидных желёз является дефицит парат-гормона и персистирующая гипокальциемия, вследствие чего развивается судорожный синдром, который может проявиться уже в первые часы жизни (неонатальная тетания). Причиной смерти детей в более старшем возрасте служат осложнения, связанные с пороками развития сердца.
Нарушения, затрагивающие Т-лимфоциты, могут быть как очень глубокими, так и едва заметными. В любом случае функция Т-клеток с возрастом восстанавливается и к 5 годам, если ребёнок остаётся жив, не удаётся обнаружить их недостаточности. Антиген-независимый этап созревания Т-клеток при этом происходит вне тимуса — в многослойных плоских эпителиях, прежде всего в эпидермисе. Одним из эффективных способов лечения синдрома Ди Джорджи является трансплантация эмбриональной ткани тимуса.
Синдром Дункана
Синдром Ду́нкана (Х-сцепленный лимфопролиферативный синдром) — иммунодефицит, характеризующийся повышенной чувствительностью к вирусу Эпштейна—Барр. Ген повышенной чувствительности к вирусу локализован в Х-хромосоме, тип наследования заболевания рецессивный, поэтому болеют мальчики. У больных, перенёсших инфекционный мононуклеоз, развиваются длительное лихорадочное состояние, лимфаденопатия (увеличение лимфатических узлов), лимфоцитоз периферической крови, гепато- и спленомегалия. Позднее формируется В-клеточная лимфома, чаще в терминальных отделах тонкой кишки, от которой больные и погибают. Летальные исходы обусловлены также деструктивным гепатитом, вызываемым вирусом Эпштейна—Барр.
Недостаточность пурин-нуклеозид-фосфорилазы
Недостаточность пурин-нуклеозид-фосфорилазы (ПНФ) наследуется по аутосомно-рецессивному типу. Дети страдают гипопластической анемией и крайне сниженной функцией Т-клеток.
Оротацидурия
Оротацидури́я — наследственное заболевание синтеза пиримидинов, которое проявляется повышенной экскрецией оротовой кислоты (оротата) с мочой, недостаточностью Т-лимфоцитов, мегалобластной анемией и задержкой умственного и физического развития. При этом заболевании снижена активность ферментов оротидил-пирофосфорилазы и оротидил-декарбоксилазы, которые преобразуют оротовую кислоту в нуклеотид-оротидин-монофосфат, необходимый для синтеза нуклеиновых кислот.
Биотин-зависимые ферментопатии
Биотин-зависимые ферментопатии также сопровождаются развитием клеточного иммунодефицита (наследственные дефекты биотинидазы и биотин-зависимых энзимов пируват-карбоксилазы и пропионат-карбоксилазы, участвующих в метаболизме аминокислот с разветвлённой цепью — валина, лейцина, изолейцина). Заболевание проявляется уже в периоде новорождённости эпизодами кетоацидоза, неврологической симптоматикой, алопецией, кожными сыпями и непереносимостью белка (рвота, мальдигестия, дегидратация). В моче содержится большое количество органических кислот. Дети отстают в физическом развитии. Из инфекционных процессов наиболее часто развиваются кандидоз и кератоконъюнктивиты. Биотин даёт хороший терапевтический эффект.